GenioWork is a powerful tool to design, compose and execute genomic workflows. In this post we show you how GenioWork can facilitate variant calling analysis.
Variant Calling is the process by which we start from sequenced data (FASTQ file) and produce a file containing the variants (VCF file).
Let’s assume that we have two FASTQ files from paired-end sequencing ERR3863191_1.fastq.gz and ERR3863191_2.fastq.gz [1] and the reference genome hg19.fa [2].
Open the GenioWork home page, click on new workflow and insert name.
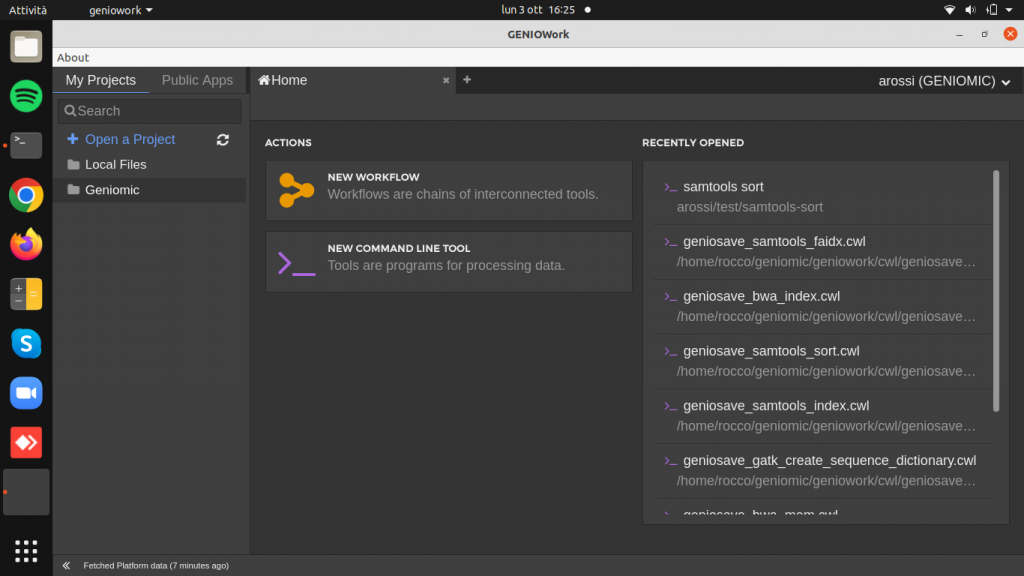
The Visual Editor page is opened: to build the workflow click on the module and drag and drop it to the design area.
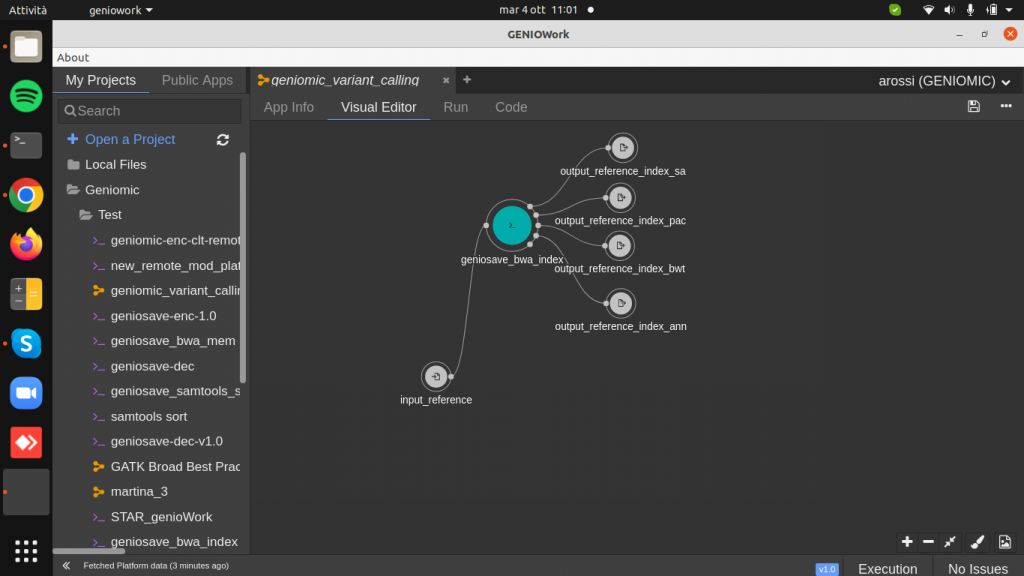
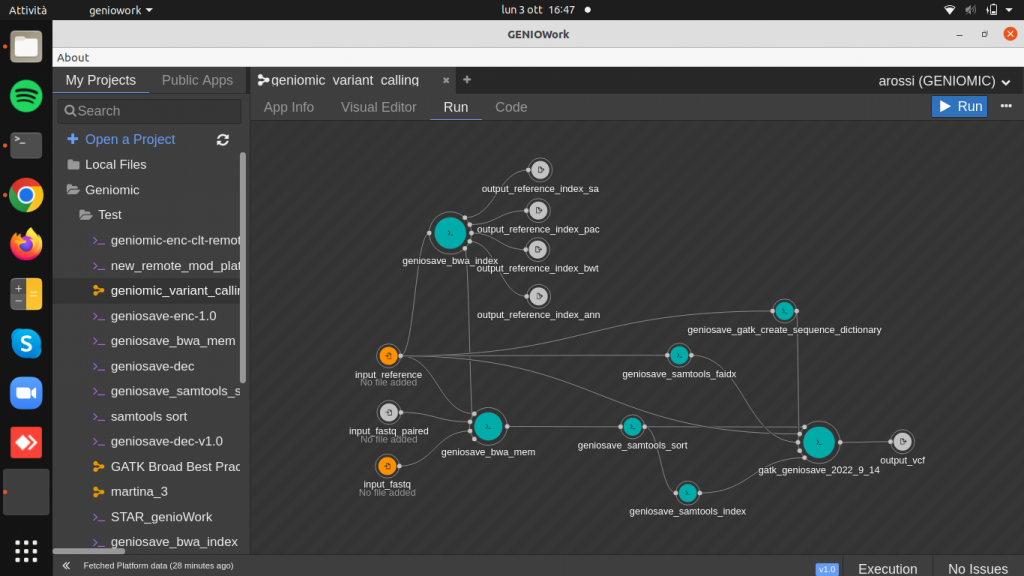
Drag and drop the geniosave_bwa_index.cwl module to the design area and create input and output ports. This module takes in input a reference genome to produce as output a reference index used to the bwa aligner.
Drag and drop the geniosave_bwa_mem.cwl module to the design area.

Clicking on the module right sidebar is opened: here it is possible to add two optional input ports, referring to the paired-end fastq file and to bwa index file and the two parameters read_group and output_sam_name.
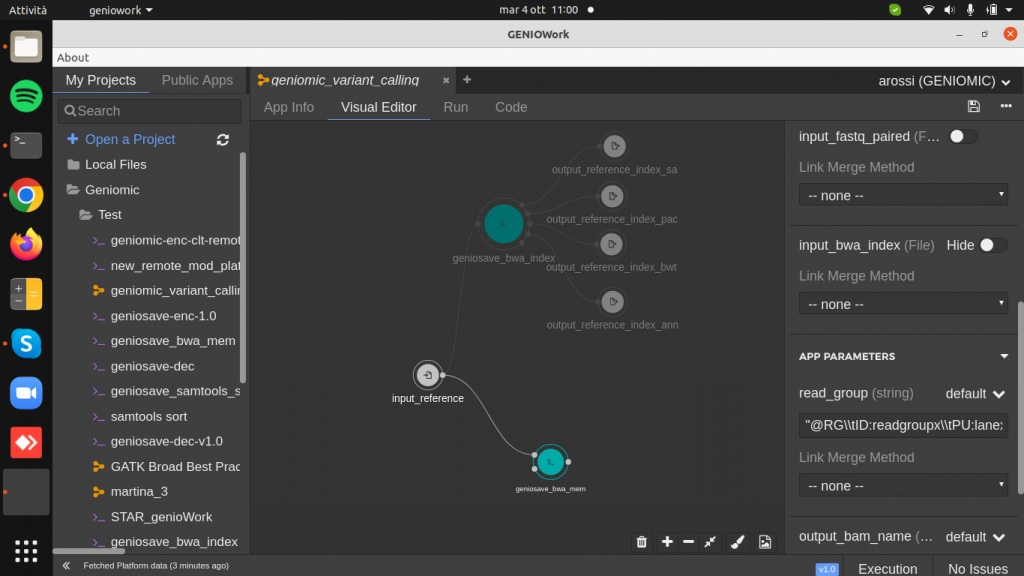
This module takes the reference genome, the index created before and the sequenced data as input and produces aligned sequence data (SAM/BAM) as output.
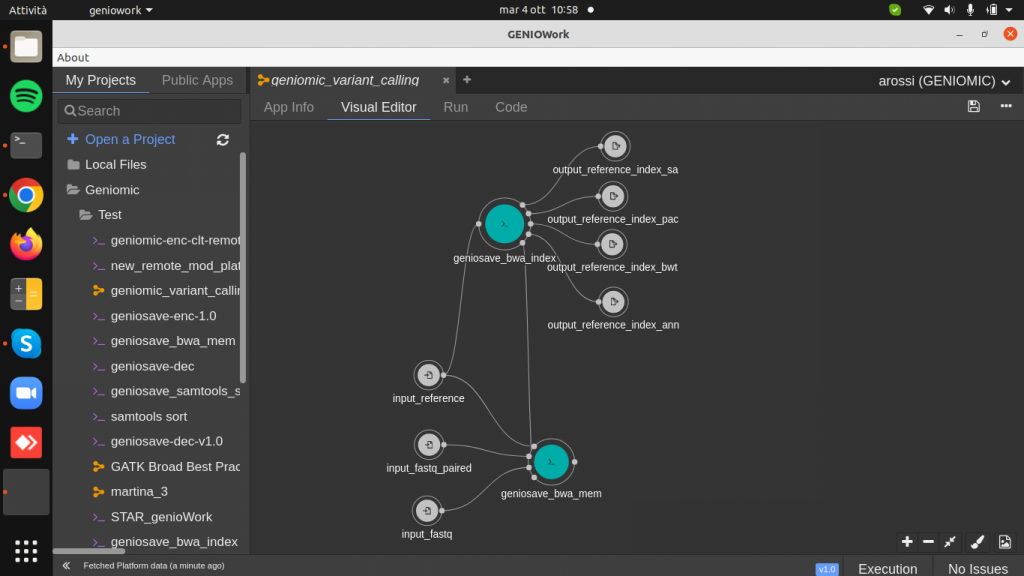
Drag and drop the geniosave_samtools_sort module (BAM sorting by coordinates) and connect the output of the aligner to the input of the samtools_sort module. By clicking on the module, it is possible to fill up the required parameter output_bam_sorted_name.
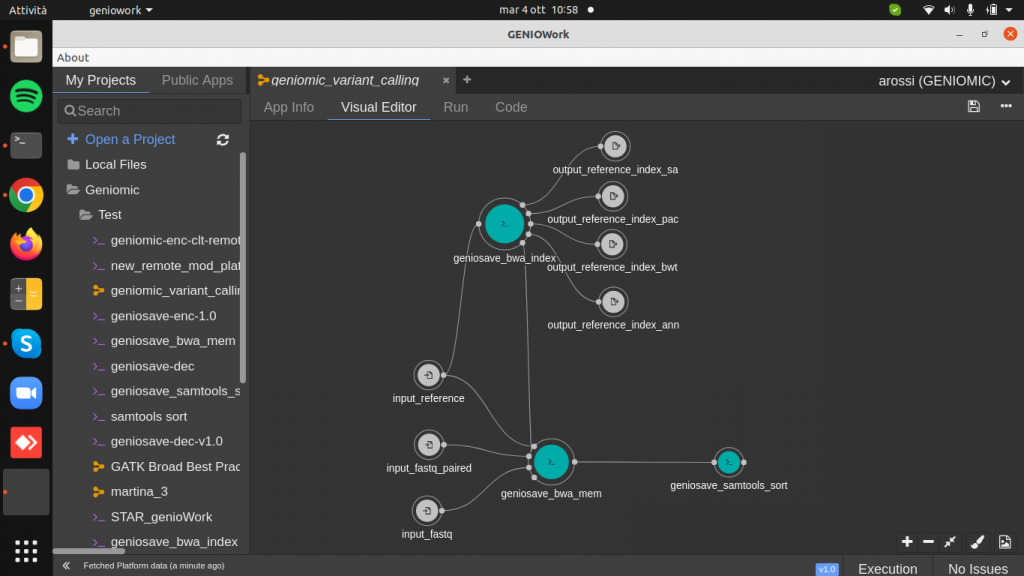
Now to identify the variants contained in the resulting BAM sorted by coordinates we go to use the geniosave_gatk_haplotype_caller module, but it requires three more secondary files:
- BAM index
- Reference genome index
- Reference genome dictionary
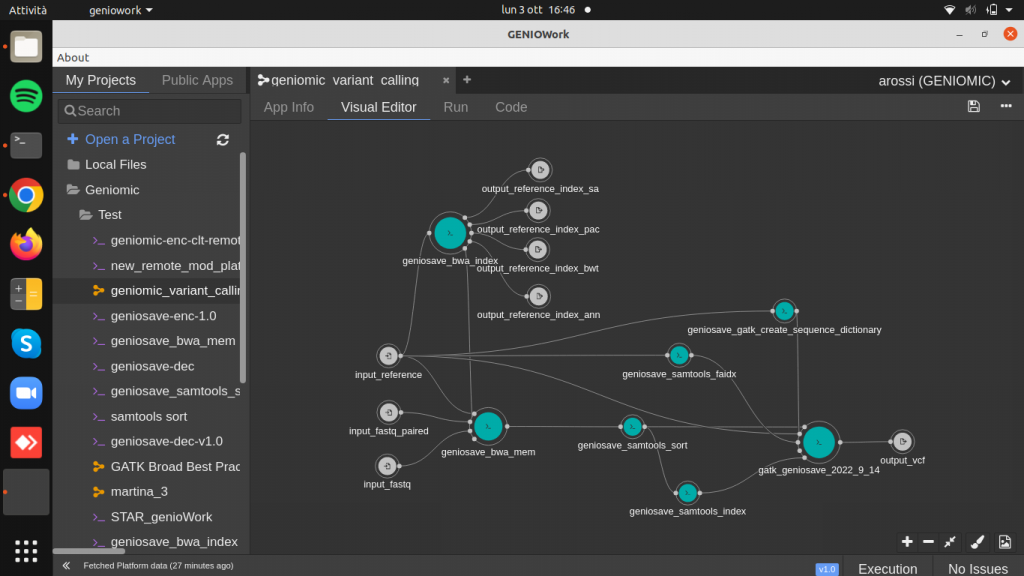
Drag and drop the geniosave_samtools_index module (BAM index) and connect the output of the samtools_sort module as input.
Drag and drop the geniosave_samtools_faidx module (reference genome index) and connect the reference genome as input.
Drag and drop the geniosave_gatk_create_sequence_dictionary module (reference genome dictionary) and connect the reference genome as input. By clicking on the module, it is possible to fill up the required parameter output_reference_dictionary_name.
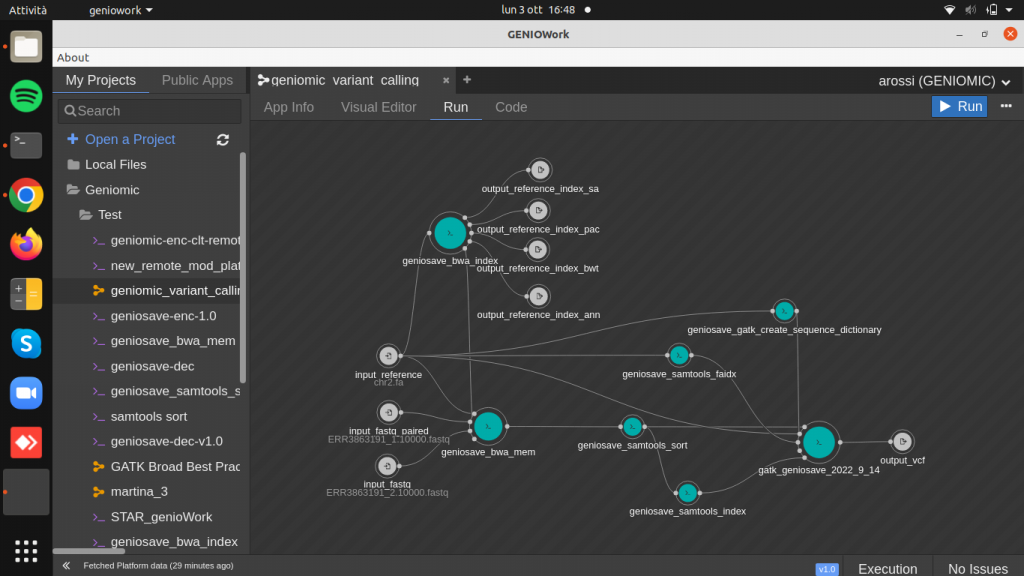
Drag and drop the geniosave_gatk_haplotype_caller (variant identifier) and connect the outputs of the geniosave_samtools_sort, geniosave_samtools_index, geniosave_samtools_faidx and geniosave_gatk_create_sequence_dictionary to the input of the geniosave_gatk_haplotype_caller.
Finally create the output port, producing the VCF file containing variants, the result of the entire workflow.
Now we pass from Visual Editor to the Test page where the input ports are filled up with the files presented in the geniomic database.
By double click on the input port it is possible to browse in the geniomic database to choose the file of interest.
The input port to mandatory fill up are represented in orange, while the optionale ones in the standard colour (white), as shown in figure.
Choose the files, clicking on the run button to run the workflow.